
Continuous Manufacturing in Pharmaceuticals: Regulatory Perspectives and Industry Insights Part 1
19 november 2024
Continuous Manufacturing (CM) is an innovative approach to drug manufacturing that applies to drug substances and drug products containing chemical entities and therapeutic proteins. It is applicable to new products (e.g., new drugs, new pharmaceutical forms or dosages, generic drugs, biosimilars) as well as to the conversion of batch manufacturing to CM for existing products (European Medicines Agency, 2023).
CM is regulated by ICH Guideline Q13, which became effective in July 2023.
CM involves the seamless supply of input materials, the transformation of materials in-process, and the simultaneous removal of output materials within a manufacturing process, without any interruption.
CM can be applied to some or to all unit operations in a process. Examples of CM modes based on the ICH Guideline Q13 (2021) include:
- A manufacturing approach in which some unit operations operate in a batch mode while others are integrated and operate in a continuous mode
- A manufacturing approach in which all unit operations of a drug substance or drug product manufacturing process are integrated and operate in a continuous mode
- A manufacturing approach in which drug substance and drug product unit operations are integrated across the boundary between drug substance and drug product to form a single CM process (i.e., the drug substance is continuously formed and processed into the drug product through integrated unit operations)
The ICH Q7 definition of a batch applies to all modes of CM. Regarding batch size, CM has flexible options as explained below:
- Batch size can be determined by run time at a defined mass flow rate, the amount produced, or raw material input. In the event of a process failure, a smaller account of the defective material can be bifurcated. This leads to less waste and a lower probability of shortages.
- Batch size can also be defined as a range. For example, defining a minimum and maximum run time can establish a batch size range (Pharma Specialists, 2023).
Real-Time Release Testing: Release testing of the drug product produced with CM is usually performed using a combination of testing on the core tablets during manufacturing (RTRT) and testing on the film-coated tablets in the quality control lab (end product testing).
Continuous Manufacturing: A Comprehensive Look at Its Pros and Cons
Continuous manufacturing is an innovative manufacturing approach that involves the continuous production of a product without interruption. It is a highly efficient process that can help businesses to reduce costs, increase productivity, and improve product quality. However, companies need to be aware of the challenges associated with continuous manufacturing
(Karkhana.io., n.d.).
Benefits of Continuous Manufacturing:
- Increased efficiency and productivity
- Improved product quality
- Improved supply chain management
Challenges of Continuous Manufacturing:
- High initial investment
- Complexityof the process
- Maintenance and upkeep
- Risk of equipment failure
- Regulatory framework is new, so authorities are still building experience and expectations and assessors’ outcome could be more unpredictable.
Figure 1
Comparison of Continuous Manufacturing and Batch Manufacturing process
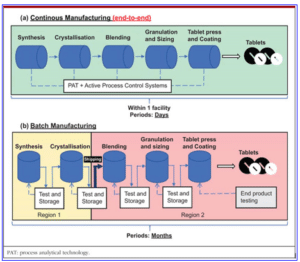
(Sia, C. H., Teh, K. S., & Chan, L. W. 2021, Figure 1)
General Implementation and Challenges
1. What are the main challenges you have encountered in implementing Continuous Manufacturing compared to traditional batch manufacturing?
- The manufacturing process is considered non-standard process if it involves partial continuous manufacturing. Therefore, process validation data should be provided in the dossier on three consecutive batches of each dosage strength at the production scale prior to authorization. The process validation batches should be manufactured using the proposed commercial running times.
- Control strategy: The relationship between end-product testing, material attributes, process monitoring, and acceptance criteria should be fully explained and justified. The use of RTRT and the relationship between the end-product should be supported by substantial comparative data at a commercial scale (parallel testing) within the current procedure.
- Batch Formula: The batch formula should be provided for the largest and smallest batch sizes. The theoretical number of tablets manufactured at minimum and maximum run time should be specified.
2. Can you provide examples of how regulatory guidelines (such as ICH Q13) have been applied in your CM projects?
According to ICH Q13 (2021), CM is defined as follows:
CM involves the continuous feeding of input materials into, the transformation of in-process materials within, and the concomitant removal of output materials from a manufacturing process.
Within Q13, there is very little guidance on process transfer and equivalency for CM processes. The guideline does address scale-up, but comes primarily from the standpoint of utilization within the same process train, or a “like-for-like.” It doesn’t really address process or equipment changes other than to say, “It may be possible.”
This creates a situation where technology users will either:
- copy and paste the same equipment, controls strategies, PAT (Process Analytical Technology), etc., which limits the use of new and improved options;
- link a process to a single process train, limiting supply chain flexibility and creating risk; or
- adopt a costly strategy for moving products within or into/out of the network, which is essentially a new process development activity (Dahlgren & Hausner, 2023).
3. Did any project face regulatory hurdles during the CM production process? If so, what were they?
No, we did not face any significant regulatory hurdles during our Continuous Manufacturing (CM) production process. We ensured that all regulatory requirements were met throughout the development and implementation phases, including comprehensive data submissions and adherence to quality guidelines.
However, it is important to note that according to an FDA statement published in February 2019 by then FDA Commissioner Scott Gottlieb, MD, and Janet Woodcock, MD, Director of the FDA’s Center for Drug Evaluation and Research (CDER), it was clear what direction the FDA is taking toward CM. The statement read:“We’re encouraged to see a growing number of companies embracing CM. It’s a key step towards promoting drug quality and improving the efficiency of pharmaceutical manufacturing. We’ve worked hard to help the industry develop the tools to start advancing these goals. The FDA is committed to helping more companies advance these CM platforms, owing to the public health benefits of these more modern approaches. We support the early adopters that are embracing this innovative technology, and we look forward to working with other interested companies.”
This reflects the FDA’s proactive stance on modernizing manufacturing processes, which aligns with our experience in CM.
4. How do you approach integrating Quality by Design (QbD) principles in Continuous Manufacturing, especially in terms of regulatory submissions?
CM can enhance the pharmaceutical manufacturing process with a magnified development approach of Quality by Design (QbD) and the utilization of Process Analytical Technology (PAT). As depicted in the figure below, a comprehensive QbD approach allows for continuous enhancement through product and process understanding to ensure better product quality (Sia, C. H., Teh, K. S., & Chan, L. W., 2021).
Figure 2
Quality by Design (QbD) Framework: Integrating Science and Risk-Based Principles
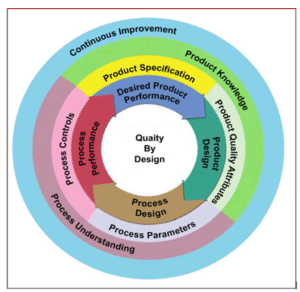
(Sia, C. H., Teh, K. S., & Chan, L. W. 2021, Figure 2)
PAT is necessary for highly automated continuous processing, as it fulfills quality requirements such as residence time distribution (RTD), and CQAs such as the percentage of active pharmaceutical ingredient (API), particle-size distribution, granule size, and many others, which can be monitored with PAT. RTD refers to the distribution of time that materials remain in a unit operation; thus, it is critical for material characterization. PAT’s primary goal is to measure critical process parameters (CPPs) that directly impact the essential quality attributes of active pharmaceutical ingredients (APIs) (Mettler Toledo, n.d.; Sia, C. H., Teh, K. S., & Chan, L. W., 2021).
PAT is an important element in a Quality by Design (QbD) approach, which supports the idea that quality cannot simply be tested in the final product but rather must be inherent by design. This approach also facilitates a smoother transition from BM to CM (Mettler Toledo, n.d.).
This article and interview were written by:
Author:
Jimmy Gandhi
CMC Specialist
ProductLife Group
Co-Authors:
Mélissa Bou Jaoudeh
Innovative Product Development Officer
Research & Innovation
ProductLife Group
Alaa Abdellatif, PhD
Innovation Product Development Officer
Research & Innovation
ProductLife Group
Supported by:
Fabio Pedna
Team Lead
ProductLife Group
The interview was conducted with and answered by Jimmy Gandhi.
Bibliography:
- Contract Pharma. (2019). Continuous manufacturing and its regulatory challenge. Contract Pharma. https://www.contractpharma.com/issues/2019-09-01/view_fda-watch/continuous-manufacturing-and-its-regulatory-challenge/?widget=listSection
- Dahlgren, G., & Hausner, D. B. (2023, July/August). ICH Q13 and what is next for continuous manufacturing. Pharmaceutical Engineering. International Society for Pharmaceutical Engineering. Retrieved October 10, 2024, from https://ispe.org/pharmaceutical-engineering/ich-q13-what-next-continuous-manufacturing
- European Medicines Agency. (2023). ICH guideline Q13 on continuous manufacturing of drug substances and drug products: Scientific guideline. Retrieved October 10, 2024, from https://www.ema.europa.eu/en/ich-guideline-q13-continuous-manufacturing-drug-substances-and-drug-products-scientific-guideline
- International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use. (2021, July). ICH harmonized tripartite guideline Q13: Continuous manufacturing of drug substances and drug products. https://www.ema.europa.eu/en/documents/scientific-guideline/ich-guideline-q13-continuous-manufacturing-drug-substances-drug-products-step-5_en.pdf
- International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (ICH). (2022). ICH Q13: Continuous manufacturing of drug substances and drug products. https://database.ich.org/sites/default/files/ICH_Q13_Step4_Guideline_2022_1116.pdf
- io. (n.d.). The pros and cons of continuous manufacturing: Benefits and challenges explained. Karkhana.io Blog. Retrieved October 10, 2024, from https://karkhana.io/blog/the-pros-and-cons-of-continuous-manufacturing-benefits-and-challenges-explained/
- Lee, S. L., O’Connor, T. F., Yang, X., & Cruz, C. N. (2015). Modernizing pharmaceutical manufacturing: From batch to continuous production. Journal of Pharmaceutical Innovation, 10(3), 191–199. https://doi.org/10.1007/s12247-015-9215-8
- Mettler Toledo. (n.d.). Process Analytical Technology (PAT) – Enhance quality and efficiency. Retrieved October 10, 2024, from https://www.mt.com/sg/en/home/applications/L1_AutoChem_Applications/L2_PAT.html
- Pharma Specialists. (2023). Continuous manufacturing of drug substances and drug products | ICH Q13 guideline. Pharma Specialists. https://www.pharmaspecialists.com/2023/03/continuous-manufacturing-of-drug.html
- Sia, C. H., Teh, K. S., & Chan, L. W. (2021). Continuous manufacturing versus batch manufacturing: Benefits, opportunities and challenges for manufacturers and regulators. Generics and Biosimilars Initiative Journal (GaBI Journal), 10(1), 44-56. https://doi.org/10.5639/gabij.2021.1001.004
- S. Food and Drug Administration. (2019, February 26). FDA statement on FDA’s modern approach to advanced pharmaceutical manufacturing. https://www.fda.gov/news-events/press-announcements/fda-statement-fdas-modern-approach-advanced-pharmaceutical-manufacturing
Register to our news and events
Go to our Events to register
Go to our News to get insights
